Our Patients
Stories that inspire
Although nano-rare patients may be one in the world, they are part of our nano-rare community. Meet these nano-rare patients and learn about their unique diagnostic journeys.
Patient stories













































































































UPDATE: Susannah received her first dose of a personalized experimental ASO medicine in October 2022. This medicine was discovered and developed for her unique gene mutation.
Antisense oligonucleotide therapy in an individual with KIF1A-associated neurological disorder
Meet Susannah
Shortly after Susannah was born, her parents noticed abnormalities in her leg movement, signaling something was extremely wrong. Their sweet little girl was later diagnosed with a frightening and debilitating nano-rare disease, KIF1A, resulting in a significant movement disorder, seizures, and speech and vision challenges. She also experiences an inescapable neuropathy in both her hands and feet that cause deep and painful burning sensations.
However, nothing can stop Susannah from shining a light on the world with her beautiful smile, positive outlook, and sincere devotion to loved ones. Just when life starts to get those around her down, she wraps her strong arms around them for a “Ginormous Suz Hug.” Those hugs are perfect. She inspires her family to live in the moment, get out of their comfort zone, sing at the top of their lungs and laugh until it hurts.

Meet Tristan
Tristan was born after a normal pregnancy in 2019. From the minute his parents saw him, they thought something looked a little off about his eyes. As none of the doctors or nurses mentioned anything, they took him home and assumed he was fine. At four months old, they started noticing some slight delays and got him into early-intervention therapies, assuming he would “catch up,” as many children do. After a year of multiple rounds of testing, they got the devastating diagnosis: a nano-rare KCNH1 genetic mutation characterized by epilepsy, profound intellectual disability, and severe speech and movement disorders. A month after Tristan was diagnosed, he had his first utterly terrifying seizure. Tristan can have seizures as a result of even the mildest illness. When he seizes, he completely stops breathing and it takes him 1-2 weeks to recover each time. The biggest impact of Tristan’s mutation is on his cognitive abilities. He can’t so much as point to something to show what he wants. His cognition was rated in the <.1 percentile with impairment so severe that many therapists have no clue how to help him.
His parents’ biggest dream is for their family to be free of the constant fear of seizures, for Tristan to be able to share what’s going on in his little head, and for him to be able to enjoy activities with his brothers rather than just watching them!

“Thanks to Lena’s acceptance into n-Lorem, hope became tangible, tangible like never before and this hope can be counted in months rather than decades. While the most experienced ASO team in the world is working on personalized treatment for Lena, we’ve also decided to add something from ourselves – we’ve opened PACS2 Research Foundation (www.pacs2research.org) with aim to understand mechanism of action of this disease and contribute to the rare-disease community.”
Meet Lena
Up until 3 months of age, Lena developed normally. She was a little bit weaker than average but being twin and premature baby was a simple explanation for this. Her family’s life changed irrevocably on the day of Christmas, 2021. Lena woke up and, out of nowhere, had her first seizure. Three days later, she was in the hospital with status epilepticus and experienced 30 grand mal seizures in 48 hours…
Despite being devastated, her parents acted fast and in Feb 2022 (thanks to DNA sequencing) she was diagnosed with a single-point mutation in the PACS2 gene, a nano-rare disease with only dozens of cases diagnosed worldwide. She struggles with epilepsy, intellectual disability, global development delay and autism spectrum.
Lena has committed countless hours towards various therapies helping her to improve on motor, social and speech skills. Despite them, she is behind in many areas, which is especially heart-breaking for her parents as they observe how she cannot keep up with her twin sister’s development and crazy ideas.
When Lena’s parents found out about n-Lorem, they knew it would be an amazing opportunity for their daughter. Putting the petal to the metal, a few weeks after Lena’s diagnosis, her application was submitted to n-Lorem by the amazing Dr. Wendy Chung from Columbia University.

Meet Sloane
While this is an incredibly challenging disorder, Sloane keeps everyone around her motivated and reminds them it’s about focusing on the now and celebrating the little wins, like recently learning to army crawl!

Meet Ireland
When Ireland was 6 months old, she had her first generalized tonic clonic seizure, which led to an epilepsy diagnosis and genetic testing. The testing revealed a CACNA1A pathogenic variant, of which the effects have been absolutely devastating. Her parents were told that no targeted treatment options were available as she was the only known case of her unique variant, classifying her as nano-rare.
Ireland presents with cerebellar ataxia, a convulsive seizure disorder with status epilepticus, developmental delays, apraxia of speech, hemiplegic migraine, and autism. Her biggest challenge is poor seizure control, despite trying a long list of AEDs, the Modified Atkins Diet, Epidiolex (CBD) and a VNS (vagus nerve stimulator) device. Ireland’s seizures are severe, never stopping without multiple rescue medications. She can completely stop breathing, requiring immediate airway intervention and support. Ireland has been intubated 9 times for status epilepticus. Between recovering from massive neurological events and frequent medication changes, Ireland often feels pretty terrible. Her family dreams of the precious day that personalized experimental ASO medicine allows her to take steps towards returning to her sweet, happy feel-good self.
Ireland works very hard in PT, OT, Speech and ABA therapies. She is able to walk, hop, climb, feed herself, and speak in single words. She continues to make developmental progress, despite setbacks caused by seizures. She has a supportive village of family and friends who dream of a treatment to reduce the heavy burden caused by her genetic variant.

“We know that Alya has so much unlocked potential with plenty to give to the world. We are deeply grateful to the n-Lorem Foundation for giving us the hope that there is a possibility, with an ASO treatment, to see Alya thrive in her life and achieve her full potential. We hope that one day Alya’s seizures are suppressed, that she’ll speak to us with real words, and that she will become independent, especially when we are no longer alive to care for her. We also have a lot of hope that Alya’s treatment will be successful, and she will be a pioneer in helping to bring this potentially life-changing treatment not only to other individuals with PACS1 syndrome but to the broader rare disease community, which is in desperate need of such treatments.”
Meet Alya
When Alya was 5 weeks old, she suddenly started to have uncontrollable seizures, one after another, and was rushed to an emergency room where she was hospitalized for a week. Initially, MRIs and genetic testing didn’t reveal anything. So, her family felt they were forced to hope that things would resolve themselves and that she would live a normal life. Her family continued to fight for a diagnosis and, at age 3, a full-exome sequencing test revealed a nano-rare disease called PACS1 syndrome. The family was told that there were no treatment options available and to do their best to manage the symptoms.
Alya was significantly delayed in everything: she smiled late, she walked late, and still, at age 9, has only 3 spoken words – for her 3 favorite people, “mama”, “papa”, and “Krish”. She continues to have significant intellectual disability, motor delays, epilepsy (even with a ketogenic diet and multiple medications), is not toilet trained, cannot feed herself, and as it stands now, she will require life-long care in the most basic areas of life (such as feeding and safety). She has been through thousands of hours of therapies, 19 electroencephalograms (EEGs) since she was born, and countless procedures. Yet, she is smiling.

“The most sincere, heartfelt thank you to all of the scientists, doctors, researchers, companies and donors that have turned a hopeless situation into so much possibility and potential for Connor, his brothers and me. And it is all happening now – in my son’s lifetime – and for that, I am eternally grateful.”
Meet Connor D.
After whole exome sequencing, he was diagnosed with a mutation in the SCN2A gene, which is the cause of all his debilitating symptoms and endless suffering. His family was told, “We don’t know much about this disease, and we have nothing new to offer you. In 100 years from now, we will have learned to silence the mutated gene, but not in your son’s lifetime”. It was a hopeless situation.
Fast forward eight years – advancing an enormous amount of research into SCN2A mutations, and Connor’s family meeting n-Lorem founder and CEO, Stan Crooke. Today, because of n-Lorem, Connor’s family is counting down the months until Connor receives his ASO, a treatment designed and tailored to his specific mutation.

Meet Margot
Margot was born healthy with no complications in 2021. At 3 months old, she started having focal seizures and needed genetic testing. The testing revealed a diagnosis of a new de novo mutation in the SCN8A gene. Margot is the only known patient with her specific mutation, making her nano-rare.
Since diagnosis, Margot has developed infantile spasms, a catastrophic type of epilepsy. She has been on 11 anti-epileptic drugs, but still has up to 80 tonic and clonic seizures a day. The SCN8A gene regulates many functions that involve the brain. As a result of her mutation, Margot’s muscles don’t work well either: she has low muscle tone and cannot control her head or sit up. She also has cortical visual impairment, meaning her brain cannot process visual input, and has trouble seeing. Margot powers through 7 hours of therapy each week to work on various skills and strengthen her body.
n-Lorem brings Margot’s family hope for their precious baby girl and for all other patients affected by nano-rare diseases.
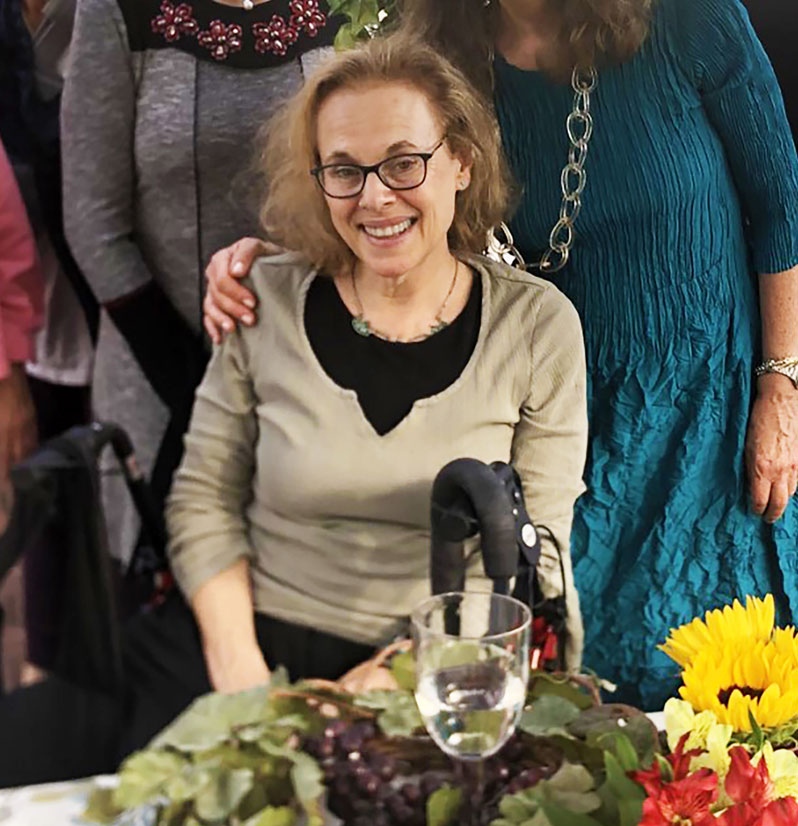
Meet Heidi
Heidi’s symptoms began 12 years ago with urinary problems. That, and the extreme fatigue she was experiencing, caused her to retire from public school teaching near the end of 2016. Her symptoms progressed to tingling and pain in the legs and feet. She consulted numerous doctors for her symptoms unsuccessfully for years before, in 2018, a neurologist finally ordered an MRI of the brain and spine. The results of the MRI (leukodystrophy, atrophy) were bewildering: abnormal, but not typical of what is seen in multiple sclerosis. Puzzled, the neurologist referred her to the Undiagnosed Diseases Program (UDP) at the National Institutes of Health.
After many tests, the UDP diagnosed her with Adult Polyglucosan Body Disease (APBD), a genetic disorder which typically manifests in one’s 50s. They told her that not only did she have this very rare disease, but that she also suffers from a nano-rare variant of the disease. There are currently no treatment options for APBD.
Heidi exists in constant, severe daily pain. She relies on a walker or wheelchair to get around the house and cannot drive anymore. Her leg strength and function continue to decline, while her pain continues to increase. She also suffers from extreme daily fatigue. She has tried numerous experimental pain treatments to no avail. Heidi has also experimented with a restrictive diet for the past two years in an effort to slow the disease progression. It is unclear whether or not the diet is effective. She voluntarily shares all of her medical data with a researcher looking for additional clues to her disease’s pathology.
Prior to the onset of APBD, Heidi was a very active and dynamic person. She was a college athlete (swimming) and continued to swim, do yoga, and exercise as an adult — until the illness made that impossible. These days, she doesn’t go out much and doesn’t see friends as she used to. An extraordinarily self-sufficient and giving person, the disease has taken away her independence and challenged her confidence. At this point, even having the energy to eat is a challenge. Once a prolific painter, she is no longer active in her studio and does not paint or draw. Playing with her grandchildren or taking them places is difficult or impossible. While she can still use her hands, she is terrified of losing that ability and more.
Heidi lives in hopes of an effective ASO treatment for APBD, one that will improve the quality of her life and the lives of others living with the disease as well. Slowing or reversing the course of the disease would change her life. Because the disease is genetic, she worries that her children or grandchildren will suffer from it as well. At this time, one of her adult children may be exhibiting early symptoms of APBD. She hopes that any effective ASO for treatment for APBD will help the many others who suffer from glycogen storage diseases.

Meet Anna
In November 2019, Anna, now 15 years old, began to speak in a strange way: After every second or third word, she took a deep breath. Her mother thought she might have developed a tic and, to be honest, she wasn’t too worried. However, since Anna constantly had severe headaches – combined with nausea and vomiting – after sports and at the end of a long school day, her mother was concerned.
The doctors consulted were faced with a puzzle: various diagnoses followed over the next few months: hormonal disorders, psychosomatic problems, allergy, myasthenia … none of them applied. Meanwhile, Anna became increasingly weak, she fell asleep at the table during the day, she could no longer hold her head up, and short distances on foot already tired her enormously.
On October 20th, 2020, the family’s world collapsed: a genetic diagnosis had been made and they received the diagnosis: amyotrophic lateral sclerosis (ALS) caused by a rare mutation in a gene that causes a severe, aggressive form of ALS. The family was devastated. Week after week, Anna lost other functions: speaking, swallowing, moving, facial expressions, gestures – everything decreased. In early November 2020, Anna underwent a percutaneous endoscopic gastrostomy (PEG), which increasingly became the only way she could feed herself. The family was desperate, but they didn’t want to give up.
Through Anna’s grandfather, a former family physician, and his friend, a professor of genetics in Newark, the family came into contact with Dr. Neil Shneider, a leading ALS researcher and physician. Dr. Shneider was instrumental in the administration of an experimental ASO that was discovered and developed for Jaci Hermstad. Jaci Hermstad was a true heroine, and despite living longer than expected, she sadly progressed from her disease in the spring of 2020. As it turned out Jaci and Anna shared the same aggressive form of ALS. As treatment was not possible in Germany, Anna and her mother flew from Germany directly to New York on December 12th, 2020, where Anna received the first dose of JaciFUSen, named after Jaci, on December 15th, 2020 at the Columbia University Irving Medical Center/The Neurological Institute of New York/New York Presbyterian Hospital. The openness, warmth and empathy that Dr. Schneider and his team welcomed them with was overwhelming. With Anna, the team felt that they had an opportunity to treat at an earlier stage in disease and that they would get the chance to do everything possible for Anna.
Anna wants to prove that not only can you survive with ALS, but you can also live a good life. She does this for herself, for her family and friends, for her beloved dog Snoopy, for Neil, for Dr. Stanley Crooke and all the doctors and therapists who believed in her,’ and especially for Jaci Hermstad, her personal heroine, whom she would have loved to meet and speak to.

Meet Mostyn
The purest example of Mostyn’s character emerged in the spring of 2021. Mostyn’s parents were taking him to quite a few baseball games. He was so excited to be back in ballparks after the loosening of Covid-19 restrictions and had become pretty much obsessed with getting a game ball. Mostyn and his parents were at a game sitting about fifteen rows above the New York Yankee’s dugout. There was another little boy and his father at the end of the row. The boy was a young teen and Mostyn was nine. The boy was very sharp, and he knew most of the players’ names in Spanish and English. He collected four baseballs by running up and down the aisle and calling for a ball each time the players came off the field. Near the end of the game, the Yankees were returning to their dugout when their first baseman, Luke Voit, threw a ball towards Mostyn. His father caught the ball and handed it to Mostyn, as the crowd let out a nice applause. Mostyn hobbled down the row toward the boy and his father. They looked at Mostyn, but he did not say anything. Mostyn took the ball out of his glove and put it in the boy’s glove. The father told the boy, “No, you cannot keep that ball” so the boy handed it back to Mostyn. But, Mostyn put the ball back in the boy’s glove before turning and walking away. The crowd erupted with emotion and many people came up to meet Mostyn at the games thereafter.
On a good day, many people would not realize Mostyn has suffered thousands of seizures. He has been on extremely high doses of at least three anticonvulsants, exclusive of rescue medications, has had multiple reconstructive surgeries on his legs and feet, has learned to walk four times, and he has a feeding tube. On a tough day, Mostyn fights as hard as he can to stay alive. He convulses over and over, his oxygen level drops, he turns blue, chokes, vomits, wakes up with ringing in his ears, loses his coordination and his ability to walk, and he gets very scared. The rescue medications compound the side effects of his daily medications, despite additional medications to counterbalance them, which have even more side effects. The medications can suppress his blood pressure, heart rate, and his ability to breathe on his own. Even slight adjustments can have major impacts on Mostyn’s emotions.
Mostyn is an only child. His parents have been extremely dedicated to his wellbeing, even prior to his birth. During pregnancy, his mother was diligent with her diet, took prenatal vitamins, avoided alcohol and fish, and drank plenty of water. Mostyn was delivered naturally with no drugs, precisely on his due date. At birth, his parents opted for optional genetic testing that was offered to them. Everything seemed to be in order and Mostyn and his mother were discharged after spending just one night in the hospital.
Mostyn was a wonderful baby. He had a good appetite and an amazing personality. He was a little slow to start walking, but his pediatrician assured his parents that everything was fine and boys sometimes take longer than girls to start walking.
When Mostyn was three, it was clear he had some developmental delays. His parents took him to a neurologist who ordered an electroencephalogram (EEG) and a brain MRI. The EEG presented some abnormal activity, but neither the MRI nor the doctor yielded any further explanations. Mostyn presented signs of cerebral palsy, hypotonia, and dyspraxia.
By the age of four, he was already in over ten hours of therapy each week: occupational, speech, and physical. Despite this, his gait was abnormal, and his wrists were weak and floppy. Kids started calling him names and making fun of him, while adults said far more hurtful things. His parents tried many things to help him like massage therapy, electrical nerve stimulation, and kinetic tape but nothing seemed to work very well. Then, one day, his father came home with a baseball glove for him, and they started playing catch every day. Beyond strengthening his wrists, Mostyn fell in love with the game of baseball, and has been wearing a baseball glove every single day for over seven years now.
Even at the age of five, speech was difficult, but he worked very hard with speech therapists to expand his vocabulary to nearly one hundred words. He was able to walk, run, climb stairs, speak, swallow, eat and drink. Then, on New Year’s Eve in 2016, Mostyn suffered an extremely violent tonic clonic seizure that lasted several minutes. Within a few weeks, he was having over thirty tonic clonic seizures a day. Mostyn’s parents took him to an emergency room, and he was later transferred to the epilepsy floor. After a couple days, a doctor came to Mostyn’s room and informed his parents that he had Lennox Gastaut Syndrome. His father asked what that meant and what his life expectancy would be. In tears, the doctor left the room. By the time Mostyn was discharged from the hospital several days later, Mostyn could no longer stand on his own. He was having tonic clonic, absence, atonic and myoclonic seizures. His parents took him home, covered all of their windows, barricaded their front door, turned off all of the lights, televisions, radios and tried to avoid any sort of stimulation, but the seizures continued. Mostyn had multiple additional EEGs, another brain MRI and multiple genetic panels performed that provided no clear answers.
His parents continued to seek the underlying cause of Mostyn’s severe refractory epilepsy, rather than settle for treatment of his symptoms. They took Mostyn to Boston Children’s Hospital, where his new neurologists were able to identify the cause of Mostyn’s epilepsy and other neurologic symptoms – a unique de novo heterozygous mutation of a gene called KCNB1. Mostyn is currently the only known case in the world with his variant, making him nano-rare. There are currently no treatment options for Mostyn’s condition, and he continues to decline. However, Mostyn and his family hope and pray that, one day, an ASO can be discovered and developed just for him.
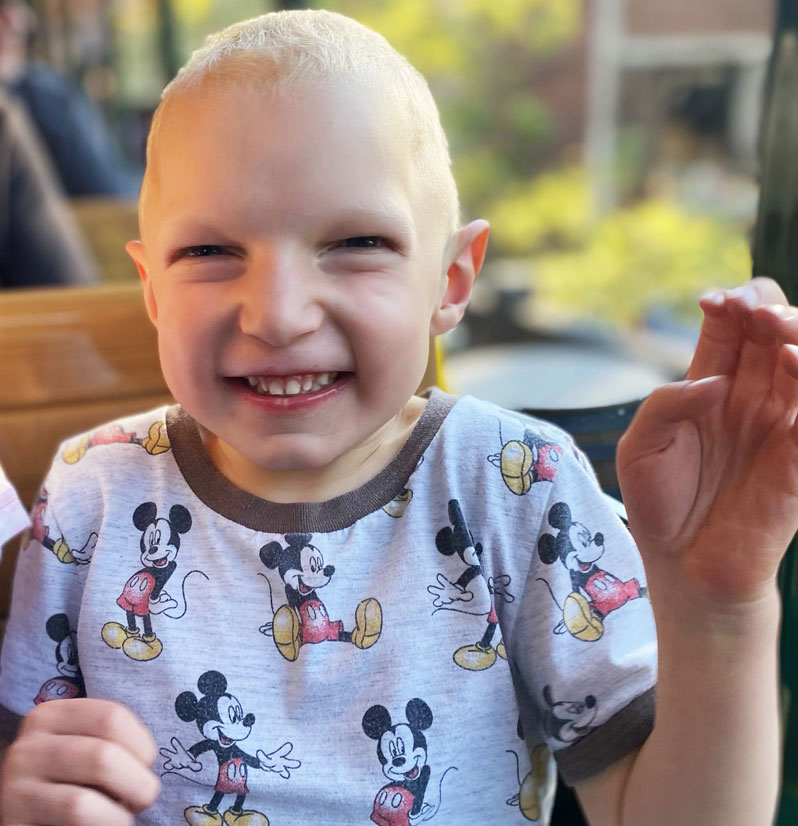
Meet Ethan

Meet Roger
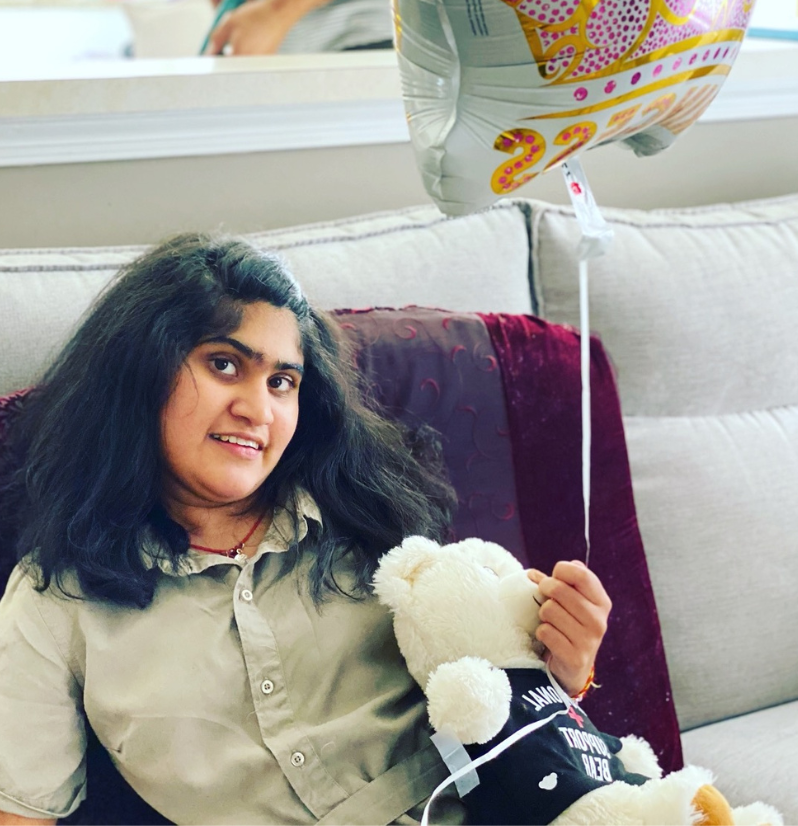
Meet Jiya

Meet Emersyn
When Emersyn was born in February 2021, she shocked everyone with her bright white head of hair and a tiny dimple/indentation on her forehead. After 20 hours with feeding difficulties, Emersyn was transferred to a children’s hospital, and then to an even bigger hospital (one we would eventually become very familiar with) and neonatal intensive care unit where she was able to get her foot in the door with her eventual geneticists. Emersyn has spent the majority of her first 2 years being poked, imaged, and tested. After two normal genetic panels, she had a whole exome sequencing test performed that revealed a CLCN7 diagnosis. Soon after, it was revealed that Emersyn is only 1 of 3 known cases in the world and, at this point, the only living case.
From November 2021 – August 2022, Emersyn spent all but 80 days (about 2 and a half months) in the hospital and multiple weeks in the ICU. Her family is thankful to have her home as much as possible. Although, they know she is extremely loved in the hospital – her second home.
Despite everything Emersyn has been through, she is extremely happy! Emersyn loves playing with her 3 older siblings on the floor and sometimes forgets that she is not an only child. Emersyn loves all things Ms. Rachel, Taylor Swift music, or anything that you can dance to.

"Our family was devastated when our son was diagnosed with a progressive neurodegenerative nano-rare disease with no cure, KIF1A.
Having a child diagnosed as nano-rare brought us great worry that research would be limited, treatment was out of reach, and Gunnar would be left behind.
n-Lorem's commitment to develop and provide treatments to nano-rare patients has brought us hope for his future. We are forever grateful to n-Lorem for giving our son the gift of potentially having a brighter tomorrow. "
Meet Gunnar
Gunnar started physical and occupational therapy, underwent serial casting, and started wearing leg braces. Despite these interventions, there was no improvement in his condition and his mobility continued to decline.
Genetic testing revealed a nano-rare mutation of Gunnar's KIF1A gene. Initially diagnosed with Spastic Diplegic Cerebral Palsy, Gunnar's diagnosis changed to KIF1A Associated Neurological Disorder and Hereditary Spastic Paraplegia.
Gunnar had surgery to reduce spasticity and orthopedic surgery to improve mobility, and he continues to work very hard to maintain his current level of ambulation. He is an ambulatory wheelchair user and enjoys playing adaptive sports – his favorite being wheelchair basketball.
Gunnar likes to make art projects and build Legos with his younger sister, Raegan. He also enjoys helping in the kitchen and baking with his mom and playing board games with his dad.
Gunnar's favorite movie is Ghostbusters. Seeing the firehouse and other locations from the movie was a dream come true for Gunnar during a trip to NYC for a KIF1A study.
Gunnar is kind, funny, and loves to crack jokes. But he's also brave and tough. Even on his worst days, if you ask him how he is doing, he will give you a thumbs up. Gunnar truly is rare and one of a kind.


Meet Talia
Talia had her first seizure at 4 months old. After a series of hospital admissions, Talia was diagnosed with Epilepsy which qualified her for rapid genetic testing. When Talia was 6 months old, she was diagnosed with a very rare genetic condition called NARS-1. There are less than 15 known cases worldwide with Talia's specific condition which causes epilepsy, global developmental delay, intellectual disability and microcephaly.
Since her diagnosis, Talia has been getting regular physio, OT, and speech support. She is very determined to learn and is working hard to grow and develop. Her persistence and focus give her parents a lot of pride as she continues to push all the boundaries.
When Talia's parents received the diagnosis, they were told effectively that there was nothing more the doctors could do, and that they had to live with the reality of the circumstance. While her parents feel incredibly lucky to have this little girl in their lives, under any circumstances, they are beyond grateful for n-Lorem which has given them hope via a pathway for Talia that may allow her to overcome her challenges with NARS-1.

"We are so grateful for the opportunity of possible treatment through n-Lorem. Knowing that someone is trying to do something for those completely unserved is a relief. We were sent home with no advice but to go home and experience a progressive neurodegenerative disease. This was absolutely crushing. To have hope for her future and possibly provide her a chance to keep singing and dancing and enjoying jokes. Nothing in this world means as much to us."
Meet Frances
Her parents started to notice that milestones were not being met and that she had some learning and speech delays, and behavioral challenges around 3 years old. Preschool was difficult for her to attend, then came the discovery she had hypotonia and tremor in her hands. In addition to this, she would also get fevers, vomit regularly and would get debilitating migraines and sleep the whole day. This was often short lived, and she would bounce back quickly, making it hard for anyone to have too much cause for concern.
Unfortunately, with these symptoms seeming mild, she was not diagnosed with a rare disease for almost 7 years. In 2021, at 9.5 years old, Frances started experiencing sudden episodes of collapsing to the ground, accompanied by fluttering eyes and slumping over. Her arms would sometimes jerk. This got progressively worse, and an EEG was done confirming myoclonic, atonic, and absence seizures. Genetic testing was advised, and she was diagnosed with a rare mutation in the DHDDS gene shortly after.

“Thanks to Kinsley’s acceptance into n-Lorem, we’ve found a sense of hope that grows stronger each day, grounded in the knowledge that the most dedicated ASO experts are crafting a personalized treatment just for her. Inspired by this hope, I, along with Daphne Graskewicz-Prado, created the Kinslow TUBB4A Foundation (www.tubb4a.org) to advocate for other families navigating TUBB4A leukodystrophy.”
Meet Kinsley
Early in her life, Kinsley’s parents noticed that she wasn’t progressing as quickly as her peers. Their first concerns were raised when, by her first birthday, she was only army crawling. In an effort to help her progress, she was placed in physical therapy which, while initially helpful, began to uncover further challenges. By 15 months, her parents noticed that she had begun to drag her toe with each step. Then by 18 months, a new symptom appeared: nystagmus, a rapid, uncontrollable movement of the eyes.
A close family friend, who also happens to be an ophthalmologist, made a few suggestions but ultimately recommended a thorough checkup. This recommendation led through a path of doctors and ultimately to an MRI to rule out a brain tumor. The results of that MRI were both a relief, in that no tumor was present, and concerning as they revealed Kinsley had less white matter than other children her age. This discovery was the springboard for a series of tests and consultations to find the right doctors, who had any answers to what was happening. In May 2023, just a few months past Kinsley’s second birthday, a diagnosis finally came: an extremely rare mutation in the TUBB4A gene. With this diagnosis came a wave of both relief and motivation for her family as it provided both answers and new challenges to navigate.
Despite these hurdles, Kinsley’s determination never wavers. She relies on a walker and orthotics making everyday movement difficult. To compound those hurdles, she is constantly fighting imbalance and difficulty with fine motor tasks. Her greatest dream is to run and play freely with her peers, but her rigid movements and equipment often make this a challenge. Kinsley’s weekly schedule is packed with therapies to help her develop skills most take for granted – from physical therapy and hippotherapy to occupational and speech therapy. Her hard work and determination are yielding results. She is working to build core strength, has learned to stand from a sitting position, and she has even learned to stand independently – albeit for brief moments. Recently, she has proudly taken her first steps and learned to undress herself, small victories that have meant the world to her and her family.
Kinsley’s family has embraced a “practice makes permanent” mentality, encouraging her to push her limits daily. Their steadfast support, combined with Kinsley’s incredible resilience, has made for a powerful force. It is this spirit of resilience and unending joy that Kinsley uses to inspire everyone who meets her, filling them with hope and happiness.

Meet Emmery
When Emmery was 9 months old, her parents started taking her to Physical Therapy because she wasn't sitting up on her own yet, and her neck wasn't quite as strong as expected. She hit most of her milestones on the later end of normal, while her twin brother was hitting them early. She quickly graduated from PT.
At 10 months, her parents noticed one of her eyes occasionally crossed, and she was prescribed glasses. She began crawling and pulling up, and her parents were so glad as they thought this was the solution to her delays.
At 16 months, Emmery saw a Developmental Specialist who suggested she was just a little behind for her age and nothing was of concern.
At 19 months old, Emmery visited a Geneticist who initially believed nothing to be wrong but decided to conduct tests for confirmation. Initial tests returned normal results, prompting a recommendation for Whole Genome Sequencing. However, the family's insurance initially refused coverage, leading to an eight-month struggle with the insurance company.
By 22 months, a Neurologist suspected a genetic disorder and supported the need for Whole Genome Sequencing. With the Neurologist's assistance, Emmery's family continued and ultimately won their fight with the insurance company. Finally, on February 8, 2022, Emmery received a diagnosis of KIF1A Associated Neurological Disorder.
Everyone agrees that Emmery is so strong, brave, and tough as she goes to multiple therapies every week and multiple doctor appointments every month. She works so hard and is extremely motivated. There's nothing better than seeing the huge smile on her face after she's accomplished a new task. Emmery is truly an inspiration to everyone.

“When we were told that two of our three children carried an ultra-rare, neurodegenerative DHDDS gene mutation, our world fell apart. The thought of having to watch not one but two of our children deteriorate physically and possibly mentally, without being able to help, was truly devastating. n-Lorem has given us hope in our darkest times—hope that a treatment pathway can be developed so that our children can enjoy the fullest life possible, and hope that they can intervene to stop this mutation from stealing away their futures.”
Meet Tom
Tom’s parents noticed that he was late with key milestones such as pointing, babbling, and clapping. When he began walking, he was clumsier than other kids and would fall more easily. Tom’s speech was also late to develop, and as a child, he would often be more interested in playing with toys than with other children.
At age four, they noticed a mild hand tremor, and when he started school, the gap between Tom and kids his age became increasingly noticeable. Tom underwent a panel of genetic testing at age ten, which came back normal. By this time, he also had diagnoses of dyspraxia and autism.
At age twelve, following growing concerns about his increasing tremor and muscle twitches, Tom had whole-genome sequencing. Fourteen months later, Tom’s family received the news that both Tom and his younger sister carried a pathogenic DHDDS mutation.


“When we were told that two of our three children carried an ultra-rare, neurodegenerative DHDDS gene mutation, our world fell apart. The thought of having to watch not one but two of our children deteriorate physically and possibly mentally, without being able to help, was truly devastating. n-Lorem has given us hope in our darkest times—hope that a treatment pathway can be developed so that our children can enjoy the fullest life possible, and hope that they can intervene to stop this mutation from stealing away their futures.”
Meet Rosie
Rosie’s parents noticed that she trembled, much like her big brother Tom, from birth. They anxiously watched her development and saw many similar delays to those her brother had encountered. Like Tom, she struggled with her motor skills and would easily trip over. While Rosie’s spoken language developed well, she struggled with her receptive language and found learning difficult. However, like Tom, it was the tremor that concerned them the most, as her little arms and hands would tremble whenever she tried to do anything.
When Rosie’s big brother Tom had his whole genome sequenced, it was suggested that Rosie be tested too. Fourteen months later, the family received the news that both of their children carried a pathogenic DHDDS gene mutation.

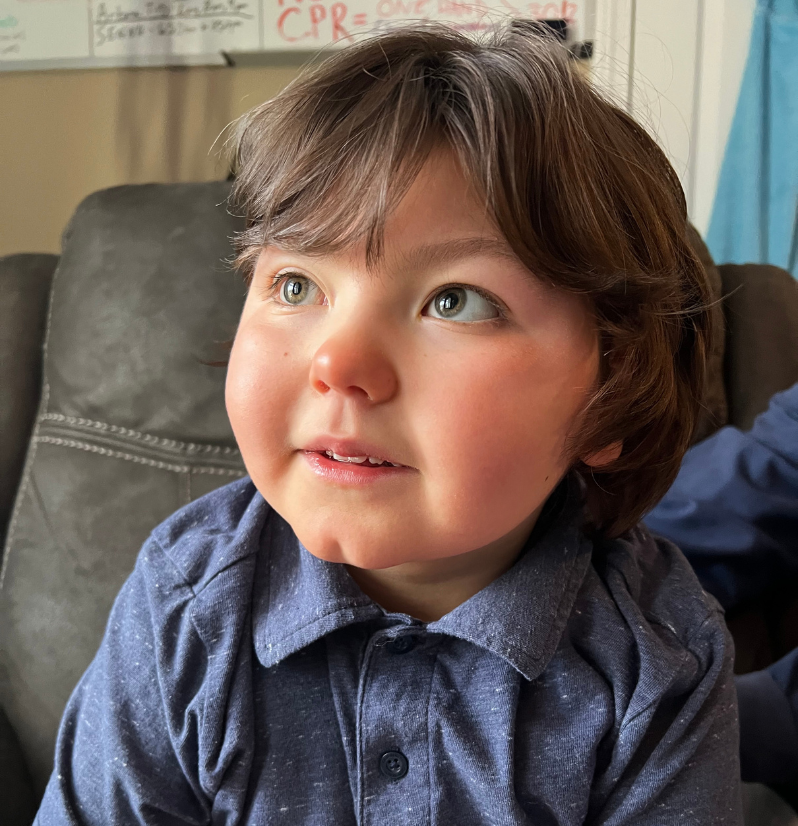
Meet Nolan
Nolan is severely disabled and globally delayed, and 100% dependent on caregivers. He is nonverbal and blind (CVI), and he also cannot independently sit up, crawl, stand, or walk. Nolan is fed through a gastrostomy tube, has active seizures and dystonia, requires frequent suctioning, and wears a CPAP nightly. Nolan is on a plethora of medications and needs frequent therapies. He is completely non-ambulatory, requiring special medical durable seating equipment and a wheelchair. His parents have been told during his numerous hospitalizations to prepare for the possibility of Nolan not going home. Fortunately, Nolan has repeatedly defied the odds and persevered, showing grit, incredible strength and a will to live.
Nolan is a happy little boy who loves his family and adventure. He enjoys his trips to the beach, mountain, or parks. Nolan prefers to be outside when the weather is just right and loves the feel of a cool breeze on his face. He has diverse musical tastes. He is cheeky, with a great sense of humor. He attends a special needs school where he has lots of friends. He loves to cuddle. But most of all, Nolan loves his big brother.
If you want to see the purest definition of love, all you need do is watch Thomas with Nolan. Thomas is Nolan’s best friend and biggest cheerleader. If you were to ask him what he would like most in the world, his reply would invariably be to make his brother better.
Nolan continues to surprise his medical team and family with his fortitude. If given the right modern treatment, no one can say what Nolan may be able to achieve. One thing is certain, however – Thomas will hold his hand every step of the way.


Meet Harlow
Known for her fierce personality, quick wit, and intelligence, Harlow embraces life with a passion that is truly inspiring. She adores her siblings — her little sister Hope and big brother Henry — and has a special love for all things pink. A true nature lover, she thrives outdoors, cherishing beach days and having a soft spot for animals. With her trusty posterior walker, she dances through life, refusing to let anything dim her sparkle.
Harlow’s journey to diagnosis began when she hadn’t started walking independently by 18 months, prompting her family’s quest for answers. An MRI revealed less than normal white matter for her age, leading to genetic testing that confirmed the TUBB4A diagnosis. Although her doctor could provide little information about this rare condition, he offered a glimmer of hope: the n-Lorem Foundation had recently visited and presented at Vanderbilt University, and he believed Harlow might qualify as a potential n-Lorem patient. A little over six months post diagnosis, her program was accepted by n-Lorem.
To know Harlow is to feel her warmth envelop you. Her heartfelt hugs serve as a reminder of the pure goodness in the world, even amid adversity. While TUBB4A leukodystrophy presents daunting challenges, Harlow’s family is committed to cherishing every moment with her, raising awareness, and finding hope in their journey together. Harlow’s spirit is unbreakable, and her story is one of love, resilience, and joy.
Every night, Harlow shares a special mantra with her mother: “Let’s Change the World Together.” With the support of n-Lorem and her unwavering spirit, Harlow plans to make a lasting impact as a beacon of hope, proving that even in the face of challenges, love and determination can light the way.



Meet Connor L.
Determined to find answers, they pursued an MRI, EEG, and extensive bloodwork. As they waited for results, they began noticing more of Connor’s tonic seizures and a series of involuntary movements that appeared seizure-like, though later doctors would rule them out as such. During this time, Connor’s developmental milestones began to slow. He stopped reaching for new skills, from rolling over to pulling up, crawling, and walking. His early babbling, once frequent and joyful, began to fade until he barely made a sound.
After many tests and consultations, Connor was finally diagnosed with a rare genetic mutation, RHOBTB2 (c.1532G>A(p.Arg511Gln)), a condition that affects only a small number of people worldwide. This mutation is known to cause neurological symptoms, including developmental delays, epilepsy, and movement disorders. Children with this mutation often experience tonic seizures, muscle stiffness, and other involuntary movements. Developmental milestones are typically significantly delayed, with many children struggling with motor skills, speech, and communication. Connor’s own symptoms—his seizures, movement difficulties, and delayed milestones—fit this profile, providing clarity on his unique needs and how best to support him.
Despite these hurdles, Connor’s spirit shines brightly. He has a few quirks that bring joy to those around him: a love for bright, colorful toys and a fascination with sensory activities, like playing with his piggy bank and opening different toys and objects. Simple joys bring him happiness, and his family finds that moments spent listening to his favorite Baby Shark music or engaging in playful interaction are some of his most cherished times. His laughter is infectious, and he has an uncanny ability to surprise his family with moments of insight and connection, reminding them of his resilience and strength.
Connor’s family continues to support him in every way possible, providing him with love, patience, and the specialized care he needs. They hold onto hope that ongoing research will lead to advancements that could improve his quality of life. His journey, though challenging, is marked by the unwavering support of his family and the dedication of a skilled medical team and therapists, all working together to help him lead a fulfilling, joyful life.

Meet Marley
When Marley was a baby, feeding challenges and growth concerns marked the beginning of her medical journey. She began facing developmental delays, leading to a diagnosis of failure to thrive and global developmental delays. Genetic testing in 2016 yielded no answers, as the NARS1 gene was not known to cause symptoms until 2020. Marley’s formal diagnosis came at the age of nine and a half, revealing she has NARS1 disorder, a nano-rare condition affecting fewer than 100 individuals worldwide. We were fortunate to find the Rory Belle Foundation, which is devoted to families affected by NARS1. Without the Rory Belle Foundation, we wouldn’t have known that this disorder causes cognitive, physical, and neurological issues, including progressive peripheral neuropathy, what research and drugs (like n-Lorem) could work, and that Marley, and we, have a community.
Marley’s biggest challenges include conversational speech, mobility challenges, due to neuropathy and low muscle tone, and navigating developmental hurdles, but her resilience shines through. She works so hard in therapies like OT, PT, speech, and ABA continuing to make progress despite her diagnosis. Marley’s newest adventure is RPM therapy (Rapid Prompting Method). It’s a way to communicate for non-speakers and unreliable speakers using letter boards, typing, and handwriting. RPM presumes competence in every student. Through this technique we are learning Marley knows how to spell complex words, can rhyme, loves poems and even knows how to do math, including multiplication. Recently when asked what her favorite thing is about herself, she spelled, “my hair.” Her parents agree!👍
Today, Marley’s loved ones are hopeful for the future where a personalized treatment will help her overcome the burden of her condition, allowing her to fully embrace her vibrant and joyful self and live her very best life possible.

Meet Colbie
Colbie’s Story: The Search for Answers
At 17½ months old, Colbie’s family received a life-changing call: after months of testing and therapies, her delays and vision concerns were linked to an ultra-rare, progressive neurological disorder called KIF1A Associated Neurological Disorder (KAND).
The diagnosis was the heartbreaking answer to several unresolved questions; from vision and balance concerns to evaluations for hypotonia, seizures, and developmental delays. Despite her challenges, Colbie continued to grow and learn with the help of therapy, even as her parents sensed something more complex was unfolding. Whole exome sequencing confirmed their instincts, revealing a rare variation in the KIF1A gene.
KAND has a wide spectrum of outcomes. While Colbie continues to make progress in some areas, her future remains uncertain. She faces the possibility of increased spasticity, vision loss, seizures, and a lifelong need for assistive devices and therapies, amongst other challenges.
Through every unknown, one thing has remained constant: Colbie’s strength and her family’s fierce love and advocacy. Their journey has been marked by heartbreak, hope, and a deep commitment to giving her the happiest life possible.
Colbie Today
Now several years past her diagnosis, Colbie continues to meet challenges that most children never have to face. She has global developmental delays impacting her both cognitively and physically. At school, she uses both her walker and wheelchair with growing confidence. She attends 4 therapies a week, takes preventative medication for epilepsy and manages ongoing vision and fine motor challenges.
Yet none of this dims her spirit. Whether making her siblings laugh, soaking up one-on-one time, or giving her all in therapy, Colbie approaches life with joy, grit, and an unstoppable will to keep moving forward. KIF1A may shape parts of her journey, but it doesn’t define her. Her light shines through in everything she does.


Meet Anais
Anais was born a seemingly healthy baby. Nothing about the pregnancy or her birth appeared out of the norm. However, as the months went on, it became clear that she was not meeting developmental milestones. She didn’t crawl until she was one, didn’t walk until she was two, and still struggles with her gait. Although she said her first words on time, she gained very few and lost them all by age four. Initially, her pediatrician was not overly concerned, so her family sought out a developmental pediatrician.
At the age of two, Anais was diagnosed with Autism and Global Developmental Delay. Genetic testing was conducted at that time, but a full genetic panel was not run. As a result, her HNRNPH2 genetic disorder was not discovered until she was six years old, when a new neurologist recommended whole exome sequencing. In the meantime, her family was left without answers as Anais continued to experience regression in skills, self-injurious behavior, severe anxiety, and a seizure that led to hospitalization.
This journey with Anais has been filled with fear and worry about her future, but her perseverance, radiant smile, and endless requests for hugs and kisses serve as daily reminders to stay positive and hopeful.


Meet Layken
A Glimmer of Hope
One of her greatest joys is spending time with her slightly younger sister, Londyn. Whether they’re swimming, playing doctor, or attending therapy sessions together, their bond is strong, and their laughter often fills the room.
Layken was born on September 8, 2016. Her entrance into the world was swift and beautiful—she earned perfect Apgar scores and looked perfectly healthy. A low body temperature at birth required some extra warming blankets, but nothing at the hospital indicated there was anything of concern. It wasn’t until the family returned home that subtle signs began to appear. Layken started losing weight, despite feeding well. Her parents had to work hard to keep her awake during feedings. Concerned, her mother raised questions with their pediatrician, only to be reassured repeatedly that some babies simply take time to catch up.
Though her instincts whispered otherwise, Layken’s mother poured her love into caregiving and tried to trust the process. But by nine months, developmental delays became more obvious. Layken struggled to sit up, showed little interest in toys, and could only engage with one at a time. Still, doctors advised waiting until she turned one.
Her first birthday came and went. Layken could sit with assistance, but she still couldn’t crawl or stand. That’s when early intervention began. She was enrolled in multiple therapies—physical, occupational, speech, feeding, and nutritional support.
After countless appointments, they were referred to a geneticist. The initial results came back inconclusive. But hope never left their side. Eventually, they found their way to Duke University, where Whole Exome Sequencing was performed. Nine months later, the answer finally arrived: Layken was diagnosed with an extremely rare neurodevelopmental disorder—HNRNPH2.
This diagnosis explained everything. From seizures and global developmental delay to hypotonia, mobility issues, language impairment, sensory processing challenges, gastrointestinal issues, CVI, and more. The list was long and the road ahead unclear, but there was finally a name for the struggle—and with that, came direction.
A new chapter began with n-Lorem, a foundation dedicated to helping those with nano-rare conditions. Layken was selected to receive an antisense oligonucleotide (ASO) therapy custom-designed for her condition. Since starting the treatment, progress has been visible, and hope continues to grow stronger.
For Layken and all of her genetic brothers and sisters battling HNRNPH2, the journey is far from over. But with ongoing support, resilience, research, and the clinical trial, the future shines brighter. Her story is one of courage, faith, and unwavering love.
After every storm, comes a rainbow—and Layken is the very definition of hope.

Meet Grace
There has never been a time in Grace’s life when she was fully healthy—not even in utero. Grace had tracheomalacia at birth, and she started showing gross motor delays by the time she was 6 months old. When Grace was a year old, her parents realized she could not feel pain after several incidents that should have been very painful. When she was nearly three years old, her family was told that she was legally blind, and by the age of five, they received a diagnosis for her condition—Posterior Column Ataxia with Retinitis Pigmentosa (PCARP), caused by a mutation in her FLVCR1 gene. Even at that young age, Grace had advanced symptoms, showing that her disease was severe.
Through the years, Grace has struggled with health issues, particularly infections. Since she cannot feel pain, infections become very severe before they are visible, resulting in more than 20 hospitalizations throughout the years. Because she is legally blind, she has difficulty assessing her body for wounds, as most people who do not feel pain learn to do. Her disease is progressive, and although she is at the end point of the sensory nerve component that makes her unable to feel pain, she still has some vision.
Several years ago, Grace’s ophthalmologist approached her family with an idea to save her remaining vision—a first-of-it’s-kind experimental ASO treatment designed specifically for Grace to stop the progression of her eye disease. Her family eagerly agreed to apply to be a candidate for an n-Lorem medicine. However, COVID intervened, and it was several years before Grace was able to receive the first dose of the experimental medication, which she received on her 13th birthday. She has received doses every six months since that day. Although it may still be too early to tell, it seems to Grace and her family that the medication is working—Grace’s vision has not worsened since that first dose.
Without the n-Lorem Foundation providing this medication to Grace, she may not have had the chance to preserve her remaining vision. Importantly, Grace is able to assess her sometimes life-threatening skin infections, which could save her life.
Grace will continue to face challenges throughout her life due to her disabilities, but her family is able to breathe a little easier knowing that both they, and others, are working to preserve her remaining vision. Grace is grateful. She is even able to read snippets of her “book children”—the hard copy versions of her audiobooks—due to the eye treatment she’s receiving. Her future is looking brighter than it ever has.


"Being a parent to a child with a disability brings both immense love and unexpected challenges. It's a reminder that any one of us could be in this position. We are not special; we're simply doing everything we can to help our child thrive. For the first time since he was diagnosed, n-Lorem is giving us hope—hope to ensure Beau has the chance to live a fuller, healthier life, one where he remains connected to the family and community he knows and loves."
Meet Beau
Beau's Diagnosis Story
After years of infertility and miscarriages, Beau was born healthy with no signs of disease. As a "COVID baby," he spent much of his early life at home, with limited time in daycare. Over time, his parents began noticing developmental delays—late crawling, poor balance, and difficulty responding to his name. At 15 months, Beau began physical therapy, and the full extent of his delays came into focus. Doctors initially considered diagnoses like autism or cerebral palsy, but genetic testing revealed something far rarer. On December 17, 2021, Beau was diagnosed with KIF1A-associated neurological disorder (KAND)—a progressive, currently incurable neurodegenerative disease. At the time, Beau was one of only a few hundred people worldwide known to have KAND. His family leaned on the KIF1A.org community for support and remain steadfast in their advocacy for treatment and research.
Life for Beau
Beau's daily life is filled with therapies, adaptive equipment, and support services designed to help him stay strong and engaged. He uses AFO braces, a walker, wheelchair, and other medical tools to move through his day safely. At school, Beau receives occupational, speech, physical, and visual therapy with the help of a 1:1 aide. Outside of school, he works hard through music, swim, horse, and neuro-based therapies. While navigating challenges like epilepsy and frequent injuries, Beau continues to explore his world with wonder and excitement. His days may require extra gear and planning, but with the love of his family and the right support, he's facing each step with courage and curiosity.



Meet Connor G
The first few months after he was born, the doctors gave him a perfect bill of health, and likewise, everything was perfect in his family’s lives. Perfect until they themselves made the heartbreaking discovery that Connor had nystagmus, a high frequency trembling of his eyes that could signal a serious neurological condition. At first, doctors told his family not to worry, that the internet is full of worst-case scenarios, and that everything would be fine.
Nevertheless, they kept pushing to learn more, and every step of the way the diagnosis worsened. As they learned the hard way, the worst-case scenario is somebody’s reality and can indeed happen to you.
They now know, as confirmed via genetic testing, that Connor has TUBB4A-related leukodystrophy, an extremely rare neurological condition which results in hypo-myelination of the brain. This condition delays and impairs the development of Connor’s motor skills and results in the slow deterioration of his nervous system.
His parents were suddenly confronted with the horror that their perfect little baby would never be able to walk, never be able to talk, and might not make it to adulthood. All of this is because of a random mutation in just one base pair of his DNA. They were devastated and heartbroken.
However, they couldn’t allow themselves to mope around. Connor is still the same happy and wonderful boy who spends all day laughing and playing as much as he can and rarely complains or cries despite the frustrations he faces. So, they continue to live life to the fullest as a happy family, trying not to dwell on the looming challenges.
Meanwhile, they are doing all they can to help support those conducting cutting-edge research that may provide a future treatment and, hopefully, help restore Connor’s future.

Meet Camille
Her family noticed that, at the age of 18 months, Camille wasn’t hitting her milestones. She began early Intervention and was making slow but steady progress. She displayed low muscle tone and had trouble with balance and coordination.
Fast forward to March of 2023, when Camille had a medical emergency that ultimately caused her to be intubated and sedated for five days at Boston Children’s Hospital. While she fully recovered from what is now known to be a hemiplegic migraine, there were no answers as to why the event happened. Boston Children’s ordered Whole Exome Sequencing, and that is when her diagnosis of a genetic mutation in the RHOBTB2 gene was made.
Her family decided from that very first day that they would not stop advocating and searching for anything that could help Camille. Eventually, they found n-Lorem, which has provided them with a sense of hope that, maybe one day, Camille will reach her full potential with the help of an ASO treatment.
While Camille still struggles with coordination, has limited expressive language, and needs help with all aspects of her day, she wakes up every day with a smile and continues to put smiles on the faces of those lucky enough to know her.

Meet Rosie
Rosie’s journey took a hopeful turn when her family discovered the YellowBrickRoad Project, a parent advocacy group dedicated to supporting those affected by HNRNPH2. They felt fortunate to connect with this incredible community of passionate families committed to making a difference.
Rosie brings joy to everyone she meets with her infectious smile and radiant spirit. Her love for music shines as she dances with exuberance, waving her “dance hands” and bouncing in her chair, filling the room with happiness. Although Rosie doesn’t have words to express herself, her eyes convey a world of emotions and thoughts, making it clear that she communicates in her own beautiful way.
However, more challenges arose around the age of three. Rosie developed serious seizures, experienced troubling regression in her development, and underwent multiple major surgeries to address her hip dysplasia. Yet in 2023, hope blossomed again when Rosie was selected to be the first HNRNPH2 patient to receive an antisense oligonucleotide (ASO) treatment through n-Lorem. With a mix of fear and hope, they embarked on this new chapter of her journey, believing in the potential for progress and better health. In September 2024, Rosie received her first dose, and her family hopes the treatment will help stop her seizures and foster greater communication and independence.

"Turner is my greatest teacher, and I thank God for the gift of our boy daily. He has taught me to be happy and joyful no matter the circumstances, to find the good in everything, and that love, joy, connection, and relationships are what life is all about. Our goal for Turner has always been to give him the very best life possible and that he is happy. I think we are doing a really good job at that. We see God's miracles woven into our lives continually. I would choose you again and again, over and over every time, Turner West. You could not be more loved."
Meet Turner
Turner's purpose is to bring joy to everyone he meets and to raise awareness for disabilities. He understands everything, just talk to him like everyone else. He is happy, resilient, amazing and so cool. He is wonderfully made. He is his family's world and so incredibly loved.
Turner lives a rare genetic condition, KIF1A Associated Neurological Disorder (KAND). He has spastic paraplegia, dystonia, developmental delays, uses a wheelchair, an eye gaze communication device, a robotic walker (affectionately named Luke) and other pieces of equipment due to living with KAND.
KAND affects every aspect of his life. Turner works 5 times harder than you doing everything that he can do. He does 5 therapies a week, has had 8 surgeries, countless tests and procedures, and takes medication daily. Turner is a super trooper rock-star. He visits Children's Hospital in Denver every 3 months to keep up with his care. His amazing team of family, friends, doctors, therapists, teachers and community are the very best! Despite all of the challenges that Turner lives with, he is so incredibly happy and present and has laughing bouts daily and, of course, listens to Luke Bryan every single day too.

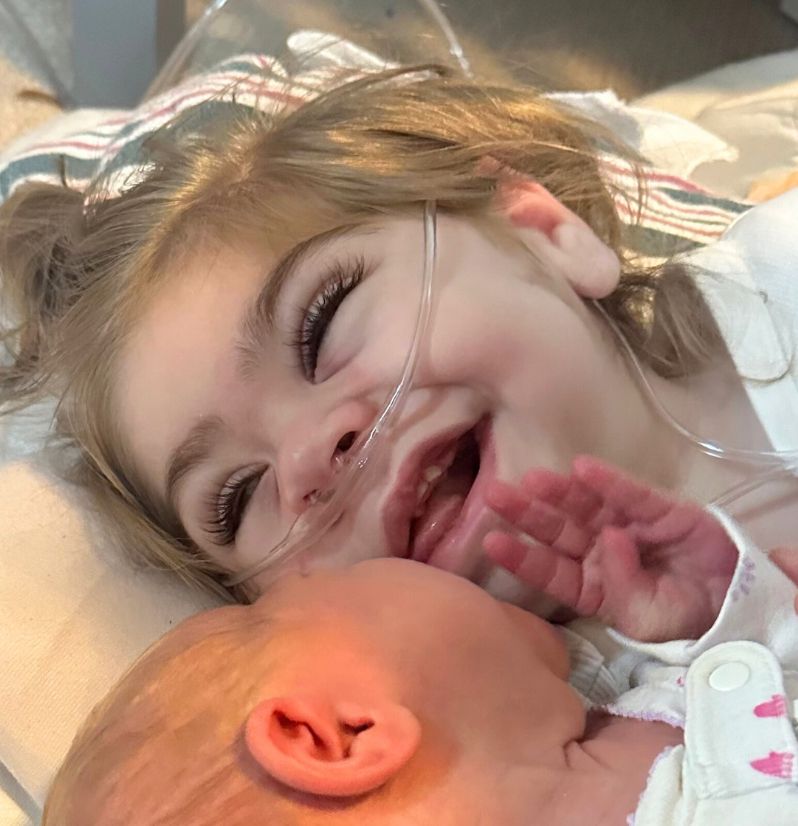
Meet Annie
When Annie was born, her parents and midwife quickly realized something was wrong – while breathing well, full term, and high APGAR scores, she wasn’t opening her eyes, crying, latching and was demonstrating a rhythmic shoulder shrug. They made the decision to transfer to the C. S. Mott Children’s Hospital at the University of Michigan, being admitted to the NICU for what would end up being a nearly 5 month stay. Mike or Emily, her parents, stayed overnight with Annie every single one of those 133 days.
Her parents and care team quickly learned that Annie was having intractable seizures. She was intubated as the team tried to stop her seizures with increasingly high doses of sedating medications, but to no luck. The MRI came back normal with no damage and because there was no known disorder or family history, the genetics team recommended genetic exome mapping, and within a week her parents learned that she had a nano-rare KCNT2 mutation. A quick google search showed just how rare this was with only ~30 known case reports in the literature at that time.
After nearly ~6 weeks of trying to navigate her feeding intolerance and as she was diagnosed with failure to thrive and severely underweight, it was clear something was not right in her GI tract. At 8 weeks old Annie had surgery for an ileostomy and G-tube. While no physical obstruction was found, it was clear Annie had GI functional dysmotility and she began taking additional medications.
Annie’s list of challenges is long – 50%+ seizure burden, abnormal background brain activity, hypotonia, GI dysfunction, neurogenic bladder, cortical vision impairment, hypoxemia, and hypercapnia.
Once Annie came home at ~5 months old, she and her family settled into a new normal where she requires 24/7 care. It has proved challenging to find sufficient home nursing support for her, often covering the night shift on top of caring for their 2 other daughters and working. Her condition is fragile with respiratory infection season especially challenging on top of her already serious condition. Adding her BiPAP machine into her care plan (however long she can tolerate before it triggers more seizures and vomiting) has been helpful to address her high levels of CO2 in her blood. Her parents and care givers have seen an increased alertness and ability to interact. This gives them greater and greater hope at the promise of a treatment through n-Lorem.
Annie’s family understands the severity of her condition and the difficult prognosis ahead and have tremendous love for her. Her ASO treatment is truly the one hope to materially change her seizure burden and improve her quality of life.
In the meantime, Annie will enjoy the snuggles of her two sisters, mom, dad and care team. She continues to have moments of alertness that allow her the simple pleasures of play, tasting yummy things (oranges are her favorite), and getting fresh air.

Meet Bowie
Following a series of genetic tests, just a week after her first birthday, they received the devastating news: Bowie had a rare leukodystrophy called TUBB4A. Her father didn’t fully understand the diagnosis at first but knew it was serious when his wife broke down in tears. Even doctors at the best hospitals knew very little about the condition—often offering quiet apologies and leaving the family grief-stricken.
Determined to give Bowie the best care possible, her parents transformed their small Manhattan apartment into something that resembled a physical therapy clinic. Though Bowie is still unable to sit up independently, when her father stands her and supports her, she proudly takes steps and beams with joy.
It took time to truly celebrate the amazing child they had. While they missed some moments trying to figure life out, their home is now filled with joy. Bowie laughs often. She loves her dog, the beach and the pool, swings at the playground, watching other children play, strolls in Central Park—and, of course, ice cream. She is bright, curious, and incredibly smart. No one has a smile like Bowie. She lights up every room she enters and has brought immeasurable light into her family’s life.

Meet Pyper
Within days of discharge, Pyper began feeding therapy, launching her family into an intensive search for the root cause of her medical challenges. After enduring six ICU hospitalizations due to pneumonia and upper respiratory infections. She was eventually diagnosed with silent aspiration, and due to treatment of this diagnosis, her infections reduced and she was accepted into an outpatient intensive feeding program at Phoenix Children’s Hospital. This program connected her family with a wide network of specialists.
Despite progress, the underlying cause of her symptoms remained unknown. With the ongoing support of her dedicated feeding occupational therapist, the family continued to search for answers. Along the way, they noticed subtle but unusual symptoms in addition to her feeding struggles: uncoordinated sneezing, sudden loss of balance, poor proprioception, sleep apnea, inability to cough, lack of tear production, absence of crying when injured, and episodes of excessive sweating. They explored every possible lead—tongue tie release, repairing a notch in her esophagus, and multiple scopes of her stomach and lungs—but none provided answers. After two years of consultations with 16 different specialists without a clear and concise diagnosis, they were referred to a genetic dermatologist due to Pyper’s unusual facial flushing. This was the first time a physician looked beyond their specialty and considered the full picture of Pyper’s medical history. Within 24 hours of that appointment, her family received a call: the physician strongly suspected Familial Dysautonomia and immediately ordered genetic testing.
One month later, the answer they had been searching for finally arrived—Pyper tested positive for Familial Dysautonomia (FD) due to a mutation in her ELP1 gene, making her the 712th person in the world to be diagnosed with this ultra-rare genetic condition. Even more uniquely, Pyper is the only known patient ever recorded to have both FD and Adrenal Insufficiency (AI). Within a week of receiving the results, they were on a plane to New York City for our first visit with the exceptional team at the NYU Dysautonomia Center, where she continues to receive expert, specialized care.
FD is a genetic disorder that affects the autonomic and sensory nervous systems, which control the body’s automatic functions. This means that many of the everyday processes most people take for granted—like regulating blood pressure, body temperature, and digestion—don’t function properly in individuals with FD.
Common features of FD include:
- Insensitivity to pain
- Unstable blood pressure and body temperature
- Absence of tears
- Poor growth
- Digestive and feeding difficulties, including the inability to suck or swallow (many individuals require feeding tubes)
- Respiratory, cardiovascular, orthopedic, and vision problems
- “Autonomic crises”—sudden episodes of severe nausea, vomiting, high blood pressure, rapid heart rate, fever, and excessive sweating
Despite the complexity of her diagnosis, Pyper faces each day with resilience, joy, and a bright spirit that inspires everyone around her. She approaches her therapies and doctor visits with eagerness and positivity. Today, Pyper continues to inspire everyone around her. Because FD affects her autonomic nervous system—including emotional regulation—she wears her heart on her sleeve, often showing her joy, love, and excitement without filter. She reminds her family daily to slow down and appreciate the small pleasures in life. Pyper’s favorite things include sushi dinners, spending time with animals (especially dogs and horses), and all things Harry Potter. Her light shines brightly, drawing people in—and leaving a lasting impression on everyone she meets.

Meet Ryker
During that initial stay at the children’s hospital, Ryker had an array of testing that would all come back normal. There were no answers as to why he was having status seizures until his epilepsy genetic panel came back. At 7 months old, he received his diagnosis of a “likely pathogenic” variant in his CACNA1A gene. Only two other individuals were diagnosed with his variant at the time. There was virtually nothing known about them or any available treatments.
Four years later in January 2021, Ryker had another life altering event and was diagnosed with severe hemiplegic migraines (HM) with brain swelling, another very rare diagnosis. His parents were so grateful that a neurologist familiar with HM was on rotation during Ryker’s stay and recognized what it was. Ryker began receiving emergency treatment for this event and was finally on a path to a slow recovery.
Over the years, many treatments were trialed. More than 10 different anti-epilepsy drugs, some off label medications, the medical ketogenic diet, and a Vagus Nerve Stimulation implant. Ryker’s CACNA1A diagnosis also comes with autism, tremor, and he suffers from debilitating ataxia that affects his balance and ability to walk independently. There are no effective treatments, and his parents focus on quality of life.
Despite all of these significant hurdles, Ryker remains a happy little boy beaming with the best smiles! He has a wonderful sense of humor with a hearty and infectious laugh. His fortitude and strength are equally matched by his resilience and ability to persevere. His love language is music that motivates nearly every aspect of his life (especially useful during therapies). He gives the most memorable deep pressure hugs. Even though he is non-verbal, Ryker has a unique ability to connect those around him and surely leaves a lasting impression.
Ryker is a gift, and his family is deeply thankful to n-Lorem for giving him a solid shot at a good, healthy life.
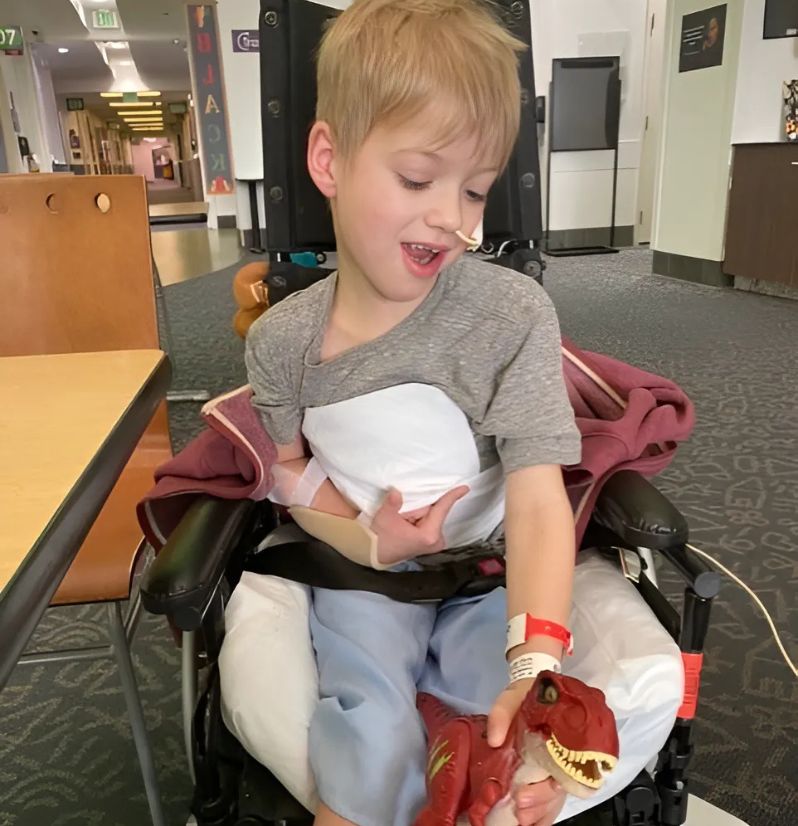
"We are so thankful that n-Lorem has taken on Charlie's program. We have so much hope for his future and that an antisense oligonucleotide (ASO) treatment will help get Charlie back to his old baseline."
Meet Charlie
Through extensive inpatient and outpatient rehabilitation therapy and medications, Charlie regained some function, but he still has no use of his right hand, has mixed tone in his right leg that affects his gait and requires him to wear braces to walk, has further reduced intelligibility of his speech, and requires a g-tube for supplementation as he fatigues when eating.
There isn't a known progression of RHOBTB2 symptoms since it is so rare. When an episode begins, Charlie loses all functionality, and the length of an episode varies, with the longest lasting multiple weeks and the shortest about 30 minutes. In the last year, the episodes have become painful and very difficult to experience. Doctors do not know if there's a possibility he won't recover from an episode or will lose more function. Also, the longer his right side is locked up, the harder it may become for him to regain use.
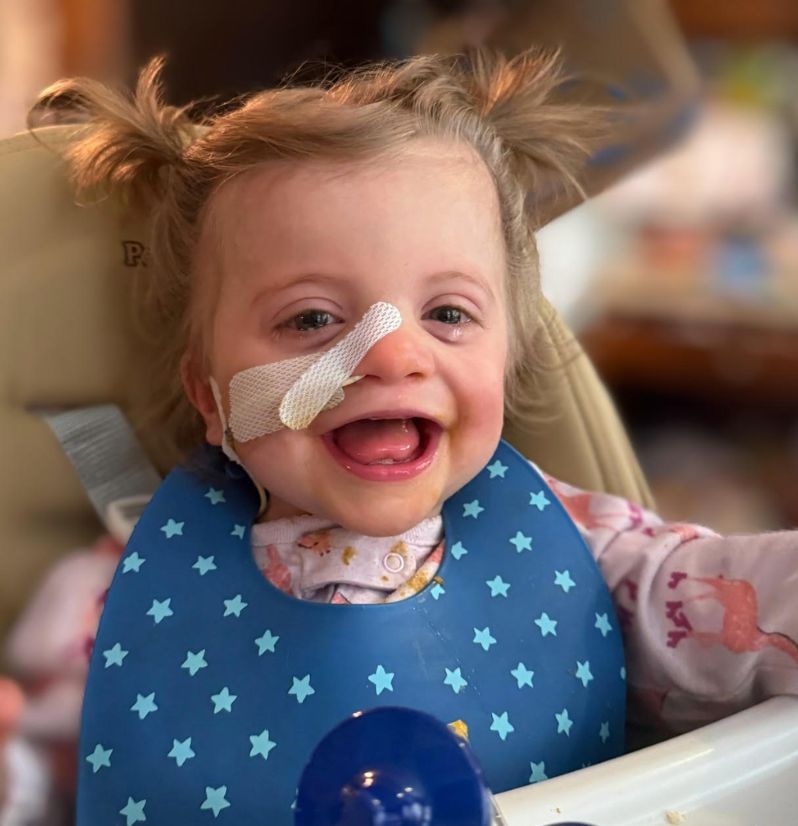
Meet Kelsey
Kelsey was born with congenital heart defects (ASD and VSD) and an irregular heartbeat. As she grew, additional challenges emerged before her formal diagnosis, including severe reflux, hypotonia, feeding difficulties, and later, cerebellar dysplasia, renal abnormalities, behavioral issues, and seizures. Persistent vomiting and oral motor challenges have made it difficult for her to eat safely, and she now receives nutrition through a feeding tube while continuing to explore food with the help of her doctors and therapists.
Despite her medical complexities, Kelsey’s determination is unwavering. She works tirelessly and has made remarkable progress; learning to sit, crawl, and play with growing independence. Nevertheless, her journey is far from over. Kelsey understands far more than she can express, and her family longs for the day when her body and mind can work together to let her share her thoughts, feelings, and imagination with the world. The n-Lorem ASO treatment offers real hope: a chance for Kelsey to continue building communication, cognition, and independence so she can more fully engage with the world around her.
Through her participation in the n-Lorem ASO program, Kelsey’s family hopes this treatment will help her reach her fullest potential and contribute to advancing understanding and care for PACS1 syndrome. Kelsey’s story is one of love, courage, and hope, and a reminder that even life’s greatest challenges can be met with laughter and an unbreakable will to keep moving forward.

Meet Tessa
Tessa is a sweet and determined little girl whose smile can brighten even the hardest of days. Born in October 2023, she was diagnosed with TUBB4A-related leukodystrophy, a rare neurological disorder that affects the white matter of the brain. Although the condition brings challenges, Tessa’s gentle spirit, resilience, and joy continue to inspire everyone who meets her.
Tessa’s journey to diagnosis began early on when she was diagnosed with low muscle tone at 12 weeks old, and soon after, her therapists observed nystagmus, a condition that causes her eyes to move involuntarily. When she still wasn’t meeting developmental milestones, her parents trusted their instincts and pushed for genetic testing. At just seven months old, they learned that Tessa had a de novo TUBB4A mutation, giving a name to what they had been seeing and allowing them to begin understanding her unique needs. While the diagnosis was heartbreaking, it also brought clarity and a path forward for her care.
Since then, Tessa has faced more obstacles than most children her age, but she approaches life with incredible strength and determination. Her days are filled with physical, occupational, and speech therapy, where she works hard on skills that many take for granted, like holding her head up, sitting with support, and practicing standing. Every bit of progress is celebrated with love, pride, and hope. Tessa’s smile melts hearts, and her spark shines brightest when she’s doing the things she loves most. She’s the happiest in the water, playing with her dog Burt, and spending time with the people she loves.
For everyone who knows her, Tessa is a daily reminder of what truly matters. Her courage has taught her family to slow down and celebrate every tiny victory, and her laughter fills their home with hope. Thanks to n-Lorem, there is now a path forward – one that gives her family renewed strength and optimism for Tessa’s future. Even on the hardest days, she continues to show that love, patience, and determination can create something beautiful: a life filled with meaning, joy, and endless love.

Meet Remy
By the age of 2 months, Remy was already showing significant global developmental delays – he lacked head control, struggled with feeding, wasn’t smiling, and didn’t visually or auditorily track. On top of that, his hypotonia, cleft palate, and trichomegaly hinted at a genetic disorder.
Remy’s parents acted swiftly and took him to see many specialists, including a clinical geneticist. At the age of 8 months, through Whole Genome Sequencing, Remy was diagnosed with DHX30 Syndrome, a neurodevelopmental disorder caused by a de novo mutation on his DHX30 gene. Most children with Remy’s missense mutation have severe motor impairment, intellectual disability, an absence of language, feeding issues, and are unable to be independent.
Despite the many challenges that life has thrown at Remy, he is a happy kid with a sweet and easygoing disposition. He has eyes bigger than his stomach, and he still hasn’t met a food he doesn’t like. He loves a live performance, and silly voices or a dance from mom and dad always bring a big, hearty laugh.
Remy is very interested in other people – especially kids – and he loves getting in close to give them a big face hug, but be careful! He might also grab your ears or mouth or nose on the way out. Remy’s first time on an adaptive swing in New York City was magical; to the delight of his parents and aunts and uncles, his laugh and shrieks of joy permeated the entire playground.
Remy keeps busy with his 13+ therapies a week. Progress is slow, but he is very motivated by bubbles, new toys, and food. Recently, he has been able to sign “more” for food and has been working hard on his “more” verbal approximation. By 18 months, he was able to roll and sit with assistance. Remy’s parents were so thrilled to be accepted into the n-Lorem pipeline and are hopeful that a personalized ASO will improve his development and quality of life.

Meet Sophie
Sophie’s first year of life was not filled with a lot of smiles or giggles though; she was a very fussy baby that always seemed distressed. As Sophie experienced lots of trouble feeding from the get-go, her family hoped things would improve as she got older. She continued to have trouble feeding and gaining weight for the first few months of life, and then her developmental delays became more evident. By the time Sophie was 8 months old, she was having trouble sitting unassisted, and she displayed some features of tone that were not expected for her age. A brain MRI shortly thereafter revealed some abnormal findings that eventually led to her NARS1 diagnosis.
Sophie started physical therapy, occupational therapy and speech before she was a year old, and she continues these therapies twice weekly and at school. Her biggest challenges continue to be communication and mobility. At this time, Sophie is non-verbal, though she uses her eyes to tell us what she wants. She practices with an augmentative and alternative communication device (AAC) and they hope one day to hear more of what she’s thinking. Sophie has recently mastered crawling, which has given her some more independence. She is working so hard in therapy to start walking. Sophie has AFOS, recently obtained a gait trainer, and she’s been practicing walking on the Locomot machine.
Her family knows Sophie will continue to need help throughout her life with the activities of daily living, but they want to do everything in their power to help her reach her fullest potential. They wish Sophie to have a happy and full life where she is content and pain-free and worry about the unknowns of her condition, especially regarding its progression and possible regression, but are optimistic for the future with the support of an n-Lorem medicine.

Meet Lincoln
Lincoln was born in September 2018, and developed normally until around 8 months old. At that time, Lincoln’s mom noticed that he was not able to sit independently on the floor without support. He often needed pillows around him because he would fall over. Eventually, Lincoln figured it out and continued to gain milestones, although slightly delayed. But at 18 months when he finally took his first independent steps, his Mom knew something was wrong. His gait was very ataxic and he looked as if he had just stumbled out of a saloon. This began a long journey of heartbreaking doctor appointments. When Lincoln was 2 ½ years old, his parents received the life-changing diagnosis of TUBB4A-Associated Leukodystrophy, a rare genetic disorder that progressively damages the nervous system.
At the time, Lincoln’s parents had no idea how this diagnosis would affect his life. Afterall, he was walking independently and was continuing to gain new skills every day. That all began to change in December 2021 when Lincoln had gotten sick with a common virus. A few days later was the last time he ever took independent steps. This is when Lincoln’s family learned that every day illnesses could accelerate decline in their son. Over the next few years Lincoln continued to lose skills he previously had—sitting independently, crawling, feeding himself, and many others. On his 6th birthday, Lincoln developed his most difficult and painful symptom yet, cervical dystonia. These intense muscle contractions pull his head and neck back against his will, making pain a part of his everyday life.
Lincoln has lost so much, but he has not lost his spark. He is one of the strongest people you will ever meet. Despite all of his physical challenges in life and constant daily pain – he never stops in his pursuit of play and laughter. After years of being told that there was nothing anyone could do, Lincoln’s parents remained hopeful and faithful in prayer. Finally, Lincoln’s parents found n-Lorem. For the first time since receiving the awful diagnosis of TUBB4A-Associated Leukodystrophy, they now have an opportunity to be excited for Lincoln’s future.

“We are forever grateful to n-Lorem and its supporters.”
Meet Amaal
Amaal’s family is hopeful that, through receiving the ASO provided by n-Lorem, she could make gains in areas that have been especially challenging for her thus far, as growing data continues to demonstrate the benefits of ASOs. Just as her name translates to “hopes and dreams,” their hope is that Amaal’s journey helps pave the way for other PACS1 families and for children across the rare disease community.

Meet Logan & Iris
Logan, affectionately known as “Lolo Monster,” is deeply curious about the world around her. She explores textures with her fingers, listens intently to voices and sounds, and is known for her high-pitched “mermaid song” when she gets tired. Iris, by contrast, is calm and gentle, often offering soft smiles and quiet babbles. She has a peaceful presence that brings comfort to those around her.
The twins spent their first eight weeks in the NICU due to feeding challenges and low muscle tone. Iris remained an additional week so doctors could place a G-tube to support her nutrition. Over the course of their first year, they failed to meet developmental milestones, and received several diagnoses including cortical visual impairment (CVI), microcephaly, epilepsy, sleep apnea, hip dysplasia, and dystonia. Eventually, whole exome sequencing revealed the underlying cause of their neurological challenges: a mutation in the SPTAN1 gene.
Logan and Iris find joy in simple, sensory experiences. They love spending time with their big brother, who showers them with affection—sometimes a bit enthusiastically. They love being close to one another, though their uncontrolled movements occasionally result in accidental face smacks if caregivers aren’t watching closely. Logan’s favorite movie is Elemental, drawn to its vivid reds and oranges. Iris is happiest upright in her stander while watching a movie or spending time outdoors. She is a true “nature girl,” captivated by the sky, a gentle breeze, or the view through tree branches overhead.
Both girls are deeply loved and supported by a devoted family of parents, grandparents, cousins, and caregivers. While little is known about the progression of their specific variant beyond early childhood, their family remains hopeful. Despite seizures that have proven difficult to control with medication, Logan and Iris’s greatest hope lies in the possibility of individualized genetic treatment and a future shaped by continued research and discovery.

Meet George
George started sitting up on his own at about one year, and around that time he started his first physical therapy intensive session. At 2 he started talking, and around 3 he was crawling on hands and knees. He is now pulling to stand, cruising, and walks with a walker. He’s done two 3-week therapy intensives a year since his diagnosis. They continue to monitor his hip development and use an abduction stander multiple times a week to aid in his bone development. He wears AFOs every day and uses an AAC device to assist with communication. In June 2025, George was accepted into the n-Lorem program.
George is a happy, social, sweet boy and his progress, while slow, has been incredible to witness. He continues to learn and develop every day, and his family is confident that he will walk independently one day. His language has exploded, and he has great receptive language. He loves music, baseball, dogs, and other kids.

Meet Janie
She used to read a lot before KIF1A affected her vision, walk briskly with canes in elementary school before a manual wheelchair became more practical, and write in a nice cursive. Now, she loves playing UNO with large cards, speed swimming in Special Olympics competitions, and rolling all over town with friends in pursuit of the best coffee. Janie regularly checks in with friends near and far and mourns too many close people who have passed over the years. Janie likes discovering new interests, like improv, and new popular songs she can sing aloud. Janie also has a serious side, wanting to protest and vote to make her opinions count. Janie says that what is important to her is having friends and support systems.
She says having KIF1A (KAND) is “very annoying” but that it is really exciting that she can hopefully get treated soon. Her parents are so appreciative of Janie’s good nature, can-do spirit, resilience when she’s feeling down, and sense of humor.
“We are hopeful this approach can help improve his seizure activity and create opportunities for developmental progress, communication, and greater engagement with the world around him. For this possibility, we are deeply grateful to Kavish’s physician and to n-Lorem. The scientists and staff at n-Lorem are bringing real hope to children like Kavish and their families through individualized treatments made possible by this groundbreaking effort.”
Meet Kavish
Kavish was born in September 2013, a fraternal twin. The earliest signs that something wasn’t right appeared when he was about two months old, when he developed significant GI discomfort and reflux. Around three months, his parents noticed tremors (rhythmic shaking) in his feet. Soon after, he began missing early developmental milestones, including eye contact and responding to sounds. At around six months of age, his first EEG showed a hypsarrhythmia-like pattern, placing him at high risk for infantile spasms, which appeared shortly afterward at about eight months old. These early events created an urgent need to find answers. When Kavish was one year old, he finally received a diagnosis: a rare mutation in the DNM1 gene. Since then, more than 100 children have been identified with pathogenic DNM1 mutations causing severe epileptic and developmental encephalopathy, yet none share Kavish’s exact variant.
This diagnosis began a long and challenging journey to manage seizures, abnormal brain activity, movement disorders, and significant developmental delays. Over the years, he has tried many anti-epileptic medications and other treatments, with varying success. Kavish remains profoundly globally delayed and is partially G-tube fed while also eating puréed foods by mouth.
And yet, Kavish truly thrives in his own way. He loves his daily massages from his mom, who was inspired by his journey to change careers and help children like him. He enjoys going out with his parents, attending school, and being part of the world around him.
An individualized ASO treatment that targets the root cause — something that once felt unimaginable — opens the possibility of new beginnings for Kavish and his family.

Meet Max
Searching for answers, they decided to pay out of pocket for Whole Genome Sequencing. Insurance refused to pay, saying a diagnosis wouldn’t affect his treatment.
Initially, the test came back negative, and Max’s neurologist said science hadn’t caught up yet. His family considers themselves as “some of the lucky ones” that didn’t have to wait much longer for a diagnosis. Two months later, they received a letter that they re-ran his sample and that he had tested positive for a mutation in the RNU4-2 gene.
They quickly searched the internet and Facebook for answers and were so glad to find others facing the same situation who knew exactly what they were going through.
While the news has brought many emotions, he continues to amaze them every single day. He is a determined, joyful, and resilient little boy—full of smiles, strength, and so much love. His resolve is a constant reminder of what truly matters, and his parents are incredibly proud.
Though their journey may look very different from what they had imagined and has not always been easy—sometimes extremely hard—they’re embracing it with optimism, courage, and a strong support system.

Meet Bill
Bill enjoys spending time with his family, hunting, fishing, and wrenching on classic cars. He is a lifetime member of the Veterans of Foreign Wars, Paralyzed Veterans of America, Disabled American Veterans and the National Rifle Association. Bill is proud to be a lifelong Philadelphia Phillies baseball fan.
Bill’s symptoms started in early 2020 when the Covid pandemic was just starting. He noticed left arm weakness and forearm pain while doing a home renovation project. During a primary care doctor visit he shared his symptoms and was referred for orthopedic and neurology consults. Knowing he had a family history of ALS he was concerned.
After months of imaging, physical therapy and repeated nerve testing, the doctors were unable to determine anything other than he had an abnormal left arm EMG test. The neurologist told Bill he did not believe he had ALS because the symptoms did not support that diagnosis. Bill decided to consult a neurologist that specialized in ALS. Unfortunately their testing and evaluation determined that he had the motor neuron disease. It took 18 months to be officially diagnosed.
Bill watched his mother and 4 of her siblings struggle and eventually lose them to ALS complications. The ALS trained neurologist recommended having a genetic test to determine if he had any of the known defective familial genes. The test identified that Bill and his family has a rare defective ALS gene CHCHD10. Bill’s daughter researched the defective gene and made contact with National Institute of Health (NIH) & Columbia University who were studying the rare gene.
After evaluation, he became a participant patient in a clinical trial called “Silence ALS”. He is being treated and studied at Columbia University Eleanor and Lou Gehrig ALS Center with Dr. Neil Shneider specifically for the nano rare defective gene. N-lorem partners with Columbia University on this trial and they developed the Antisense Oligonucleotide (ASO).
There are less than 15 patients receiving the CHCHD10-001 ASO. This treatment gives hope toward slowing of function decline and potentially future cure. This future would not exist without n-lorem’s life changing work. Bill and his family are encouraged that this treatment will be a game changer to future generations of families with our defective gene. Bill has been receiving the ASO treatments since January 2025 and they have been well tolerated with no detected decline in breathing or physical function.

“Unless someone like you cares a whole awful lot, nothing is going to get better, it’s not.”
Meet Lucas
At just two months old, Lucas was diagnosed with a CYFIP2 related disorder after experiencing his first seizure. Prior to this, there were no signs other than some mild feeding issues. He and his family were led to many specialists, but most doctors had never encountered another patient with a CYFIP2 condition, and any available research was still in very early stages.
Despite the long list of challenges caused by his CYFIP2 related disorder, Lucas remains determined and resilient. It is amazing to see him gain skills and abilities that were once thought to be impossible. He loves watching The Lorax, swimming in the pool, rolling around with Dad, and playing with his light up toys! He is extremely smart and curious. While his condition affects many aspects of his development, Lucas constantly finds new ways to learn, communicate, and engage with the world around him. Without ever having to say a word, his funny personality is hard to hide.
Lucas has taught those who care for him more about perseverance, patience, and unconditional love than ever thought possible. Upon receiving his diagnosis, his family was told “the science just isn’t there yet.” Today, they say, “There is so much hope for the work n-Lorem is doing to advance the possibilities for Lucas, within our lifetime.”

“Progress isn’t measured by how fast Jeffrey reaches each milestone, but by the resilience, love, and hope he inspires every single day.”
Meet Jeffrey
Jeffrey’s pregnancy and birth were completely uneventful, and everything seemed to be off to a healthy start. Around three months of age, however, his parents began noticing feeding difficulties, poor weight gain, and delays in reaching developmental milestones. What followed was a journey filled with medical appointments, therapies, and countless tests as they searched for answers.
In February 2025, whole genome sequencing identified a pathogenic RNU4-2 variant, leading to a diagnosis of ReNU syndrome. While it was initially overwhelming, it also brought clarity. It connected Jeffrey’s family with specialists who understood his condition and introduced them to a community of researchers, clinicians, and families working together toward better treatments. For the first time, there was not only an answer—but also hope.
Jeffrey continues to reach milestones in his own time. He relies primarily on G-tube feeding and is still nonverbal, but he communicates through his bright smile, expressive eyes, and laughter. Jeffrey has taught his family to see life through a different lens—to cherish every small step and find joy and hope in moments that others might take for granted.
They are deeply grateful to the physicians, therapists, researchers, and organizations like the n-Lorem Foundation, whose dedication continues to bring hope to families living with nano-rare diseases.
By sharing Jeffrey’s story, his family hopes to raise awareness of ReNU syndrome, encourage other families facing similar challenges. They remain hopeful that, through science, collaboration, and compassion, effective treatments will become available—not only for Jeffrey, but for every child living with ReNU syndrome.




We welcome your family
to our community
We understand that being nano-rare often means that each patient is alone in their unique disease, alone in their suffering and isolated from the healthcare community. We want to change this by empowering our nano-rare patients.
The Patient’s Journey
Developing a personalized ASO treatment plan
At n-Lorem, our mission is to treat the patients we can today. By working closely with our research physicians and their institutions, we discover, develop and provide personalized experimental ASO medicines to nano-rare patients for free, for life. This journey begins with a research physician submitting an application to treat for their patient to n-Lorem.

Find out if you
qualify for treatment
Nano-rare describes a patient that, because of their unique gene mutation, will be only 1 to 30 patients worldwide with that exact mutation. Most nano-rare patients have a single gene mutation that causes a cluster of symptoms that can affect their health. Many of these single gene mutations lead to degenerative conditions or even death.

We cannot do
this alone
Together we are changing the world—
one patient at a time
We hope that you join us on this journey to discover, develop and provide individualized antisense medicines for free for life for nano-rare patients. The ultimate personalized medicine approach – for free, for life.

We need your support